
Page 152 - 南京医科大学学报自然科学版
P. 152
第43卷第7期
·1038 · 南 京 医 科 大 学 学 报 2023年7月
[5]
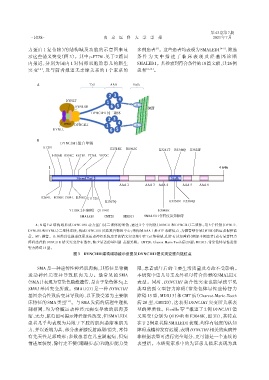
力蛋白 1 复合物)的结构域及功能的示意图来显 多例患者 。这些患者均表现为SMALED1 [4-5] ,筛选
示这些错义突变(图 3)。其中 p.P776L 见于 2 篇国 条 件 为 文 中 描 述 了 临 床 表 现 及 经 基 因 诊 断
内报道,分别为国内 1 对同卵双胞胎患儿的新生 SMALED1。共检索到符合条件的18篇文献,共26例
突 变 [4] ,及与前者报道无亲缘关系的 1 个家系的 患者 [6-12] 。
A Tail AAA Stalk
DYNLT
MT
DYNLRB
DYNC1H1同二聚体
DYNC1L1
DYNLL
B
DYNC1H1蛋白单链
K1291
E1518K R1962C
K3241T R3344Q H3822P
H306R R598C K671E P776L Y970C
1 4 646
Stem((Tail)) Stalk
AAA 1 AAA 3 AAA 4 AAA 5 AAA 6
R264L H306R 1584L R598C G1132E
R1567Q K3336N R3384Q
Y1109C(本案例) Q1194R E3048K
SMALED1 CMT20 MRD13 SMALED1伴轻度认知障碍
A:N 端 Tail 结构域形成 DYNC1H1 动力蛋白同二聚体的重链,通过 2 个中间链(DYNC1I 和 DYNC1LI)二聚体,与 3 个轻链(DYNLT、
DYNLRB 和 DYNLL)二聚体连接,构成 DYNC1H1 功能复合物的中心;结构域 AAA 1 是 ATP 水解位点,为微管结合域(MTBD)的运动提供能
量。MT:微管。B:虽然以往报道仅累及运动神经系统患者的错义突变集中在 Tail 结构域,但伴有认知障碍(例如本例患者)或有显著智力
障碍患者的 DYNC1H1 错义突变分布散在,缺少显著的基因型⁃表型关联。CMT20:Charcot⁃Marie⁃Tooth 病 20 型;MRD13:常染色体显性遗传
智力障碍 13 型。
图3 DYNC1H1结构域功能示意图及DYNC1H1错义突变蛋白链位点
SMA是一种遗传性神经肌肉病,其特征是脊髓 限,患者成年后的主要生活质量及寿命不受影响。
运动神经元变异导致肌肉无力。最常见的 SMA 本研究中患儿母亲及外祖母符合典型的SMALED1
(SMA1)表现为常染色隐性遗传,是由于染色体5q上 表型。同时,DYNC1H1 杂合性突变也能导致罕见
SMN1 基因突变所致。SMALED1 是一种 DYNC1H1 类型的孤立型智力障碍(常染色体显性遗传智力
基因杂合性致病变异导致的、以下肢受累为主要临 障碍 13 型,MDR13)和 CMT 病(Charcot⁃Marie⁃Tooth
床特征的 SMA 类型 [13] 。与 SMA 发病的病理生理机 病 20 型,CMT20),这表明 DYNC1H1 突变所关联表
制相同,均为脊髓运动神经元病变导致的肌肉萎 型的异质性。Fiorillo 等 [14] 报道了 2 例 DYNC1H1 错
缩、无力,肌电图可提示神经源性改变,但SMALED1 义突变(分别为 Q1194R 和 E3048K,图 3B),其特点
患者几乎均表现为局限于下肢的肌肉萎缩和肌无 在于2例患者除SMALED1表现,均伴有轻度的认知
力,并以近端为甚,部分患者膝反射减弱/消失,常伴 障碍或精神发育迟缓,表明DYNC1H1相关的疾病并
有先天性足部畸形;多数患者在儿童期起病,但病 非根据表型可进行完全划分,更可能是一个连续的
情进展较慢,除行走不便(蹒跚步态)和跑步能力受 表型谱。本研究家系中的先证患儿临床表现为典