
Page 146 - 南京医科大学学报自然科学版
P. 146
第41卷第11期
·1708 · 南 京 医 科 大 学 学 报 2021年11月
表1 CPT1A基因外显子4~5 qPCR扩增引物设计
引物名称 碱基序列(5′→3′) 退火温度(℃) 片段大小(bp)
ALB⁃QF上游 AGTGCACTTGTTGAGCTCGTG 60 128
ALB⁃QR下游 GCAAAGCAGGTCTCCTTATCG
CPT1A⁃4Q上游 ATCCAGATCTTGGTGGCACG 60 162
CPT1A⁃4Q下游 GTCCAGCCAGACGAAGAACG
CPT1A⁃5Q上游 AAGCCTTAATCCACAGATGGCT 60 130
CPT1A⁃5Q下游 AAACCCATGTTGTACAGCTTCC
次报道,具有种族特异性,在阿拉斯加、加拿大、格
陵兰岛和西伯利亚东北地区发病率较高 [3-5] ,其他地
区较罕见,目前中国人群的患病率未知。
CPT1A 缺陷患者临床表现差异较大,在正常生
理情况下没有明显的临床表现,当糖原储备显著减
A B 少时,出现低血糖、呕吐、腹泻、抽搐、肝肿大、肝性
A:肝细胞肿胀,胞质空亮,广泛脂肪变性(×200);B:汇管区纤 脑病等典型的临床特征 ,一些个体可出现肾小管
[6]
维组织中度增生伴少量淋巴细胞浸润(×100)。
性酸中毒表型 [7-8] 。也有个案报道以黄疸为首发症
图1 先证者肝脏穿刺活检组织光镜病理
状 。心脏或骨骼肌受累并不常见 。实验室检查
[9]
[1]
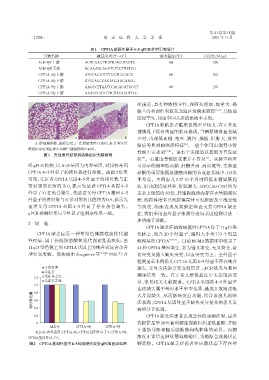
组qPCR检测,以ALB基因为内参基因,对目标基因 可显示低酮型低血糖、肝酶升高、高血氨等,实验室
CPT1A 4~5外显子的拷贝数进行检测。由图2结果 诊断的重要依据是脂酰肉碱的合成显著减少,C0水
可见,先证者 CPT1A 基因 4~5 外显子的拷贝数与正 平升高。本例患儿 2 岁 10 个月时因低血糖晕厥起
常对照的比值约为 0,提示先证者 CPT1A 基因 4~5 病,肝功能持续异常,肝脏肿大,C0/(C16+C18)约为
外显子存在纯合缺失,先证者父母 CPT1A 基因 4~5 正常上限值的50倍,肝细胞胞质内存在大量脂滴沉
外显子的拷贝数与正常对照的比值约为0.5,提示先 积,线粒体增多且线粒体局灶可见肿胀及空泡变性
证者父母 CPT1A 基因 4~5 外显子存在杂合缺失。 等改变,临床表现及实验室检查支持 CPT1A 缺乏
qPCR检测结果与全外显子组测序结果一致。 症,我们采用全外显子组测序进行基因检测以进一
步明确了诊断。
2 讨 论
CPT1A缺乏症的致病基因CPT1A位于11q13染
CPT1A 缺乏症是一种常染色体隐性遗传代谢 色体上,包含 20 个外显子,编码大小为 773 个氨基
性疾病,属于长链脂肪酸氧化代谢紊乱类疾病,由 酸残基的CPT1A [10-11] 。目前HGMD数据库中收录了
11q13染色体上的CPT1A基因上的纯合或复合杂合 41种CPT1A基因突变,多为错义突变、无义突变、剪
[2]
型突变所致。该疾病由 Bougnères 等 于 1981 年首 切突变及插入缺失突变,以点突变为主。全外显子
检测显示本例患儿CPT1A基因4~5外显子存在纯合
正常对照 缺失,父母为该杂合突变携带者,qPCR 结果与基因
先证者
先证者之父 测序结果一致。在正常人群数据库中未发现该变
1.2
先证者之母 异,未见相关文献报道。CPT1A 基因的 4~5 外显子
1.0
连续缺失属于基因水平中型变异,造成开放阅读框
0.8
相对表达量 0.6 大片段缺失,从而影响蛋白功能,结合该患儿的临
床表现,CPT1A基因外显子缺失变异是本例患儿发
0.4
CPT1A 缺乏症患者出现急性低血糖症时,应尽
0.2 病的分子机制。
0 快提供足量10%葡萄糖注射液以纠正低血糖,并防
ALB⁃Q CPT1A⁃4Q CPT1A⁃5Q
止脂肪分解和随后脂肪酸向线粒体的动员。血糖
ALB⁃Q:内参基因;CPT1A⁃4Q:CPT1A基因外显子4;CPT1A⁃5Q:
CPT1A基因外显子5。 浓度正常后应继续葡萄糖输注,为糖原合成提供足
图2 CPT1A基因外显子4~5纯合缺失突变qPCR验证结果 够底物。CPT1A 缺乏症患者在应激状态下存在神